

Allyson Lock
New Zealand Pompe Network
 Allyson, President of NZPN, opened the conference and thanked everyone for coming. She introduced the committee members and the process that had been gone through to make it all happen.
Allyson, President of NZPN, opened the conference and thanked everyone for coming. She introduced the committee members and the process that had been gone through to make it all happen.
Attendees included 8 of 11 Kiwi patients and their families, pharmaceutical companies investing in new therapies and producing current enzyme replacement therapies (ERT), and top researchers in the field from USA and Australia, along with support organisations such as NZORD and MDANZ.





















Dr Collette Bromhead
NZ Organisation for Rare Disorders
 The first guest speaker was Dr Collette Bromhead, CEO of NZORD, who talked about the work that NZORD does and how it works with patients for positive outcomes.
The first guest speaker was Dr Collette Bromhead, CEO of NZORD, who talked about the work that NZORD does and how it works with patients for positive outcomes.
Dr Bromhead stated that NZ has the highest density of registered charities in the western world, so everyone is competing for funding. Just some of NZORD’s outputs include; advocacy and profile raising with the Ministry of Health/PHARMAC/external stakeholders, hosting websites for 40 support groups, responding to patient and clinician enquiries and maintaining a specialist database. She also spoke about the briefing that was produced for the incoming Minister of Health and gave a breakdown on the medicines funded under the rare umbrella by PHARMAC, noting that the trial by PHARMAC for funding rare diseases had truly been a failure. Next up was the bombshell that funding from the government for the organization could be cut by June 2018 which will impact hugely on NZORDs ability to help the rare disease community.
The plans of what NZORD intends to do if funding is secured were laid out and included:
– Greater coordination for support groups and patient get togethers
– Offering web tools such as video conferencing to facilitate small groups to get together
– Expanding the Specialists’ Database to allied health practitioners
– Continuing to drive the agenda with Government around equitable access to rare disease medicines, a rare disease register
– Facilitating and coordinating research and clinical trials using patient-driven databases
– Hosting a site for NZ researchers on NZORD website to enhance collaboration
Dr Bromhead also focused on the NZORD Rare Disease Day symposium and poster child campaign which was designed to raise awareness of rare diseases for GPs, nurses and researchers.
A request for stories from the rare disease community was made to enable the collective voice to be heard through sharing real people’s challenges and triumphs. This ‘stories project’ can give the public and politicians insights into the real issues that are faced over and over again. The message that “together we are strong” must mean speaking up and coming together to raise the voice of the rare community enough so that decision makers can hear us!




























Dr Barry Byrne
University of Florida
 Dr Barry Byrne from the University of Florida gave a great talk on current research and newborn screening for Pompe disease.
Dr Barry Byrne from the University of Florida gave a great talk on current research and newborn screening for Pompe disease.
Information included brain involvement through accumulation of glycogen in the brain, which consequently causes damage to neurones. He emphasised that early treatment was vital to long-term positive outcomes. Interestingly, Dr Byrne stated that oestrogen appears to be protective to neurone loss.
Dr Byrne spoke about the newborn screening program in America and how they have looked to the successful model that has been run in Taiwan. This is of interest to us here in New Zealand as we are focusing on getting Pompe disease added to the newborn screening panel here.
Diagnosis of Pompe includes establishing low functional GAA enzyme levels and genotyping to rule out pseudo deficiency, identify carriers, predict infantile-onset versus late-onset, and predict Cross-Reacting Immunologic material (CRIM) status. The current treatment is enzyme replacement therapy (ERT) that is initiated as soon as possible, and early initiation of ERT improves intermediate term outcomes.
Taiwan currently screens for Pompe disease and both Washington State and Missouri are doing pilot studies on Pompe screening. Taiwan has screened 470,000 infants for Pompe and 10 infants have been identified by the screening and started early treatment. Washington has identified 4 infants with Pompe, 4 carriers, 3 carrier/pseudo deficiency, and 6 heterozygous for pseudo deficiency. Missouri has screen 43,701 samples with 27 identified to have some type of LSD, 18 screened positive for Pompe, and 8 identified with Pompe (3 infantile, 3 late-onset, and 2 of unknown significance).
Laura Byrne
 Laura provided a briefing on the benefits of nutrition and exercise in Pompe patients. She used videos of patients undertaking different exercises that were created specifically for Pompe patients. She also stressed the dangers of over-exercising, as Pompe patients need to maintain some muscle resistance to retain the ability to stay mobile.
Laura provided a briefing on the benefits of nutrition and exercise in Pompe patients. She used videos of patients undertaking different exercises that were created specifically for Pompe patients. She also stressed the dangers of over-exercising, as Pompe patients need to maintain some muscle resistance to retain the ability to stay mobile.
Laura spoke about the importance of a low carbohydrate diet and sub-maximal exercise, as much as you can when you have Pompe. She screened some slides of various exercises that mobile Pompe patients can do. There are exercises on the web that wheelchair bound people can do.
Laura talked on Pilates being positive, but Yoga stretches the ligaments too much which is bad since we don’t have the muscles to hold everything together and it causes over extension.


Freda Evans
 Freda talked about her journey from being diagnosed almost 30 years ago, to finally getting access to treatment.
Freda talked about her journey from being diagnosed almost 30 years ago, to finally getting access to treatment.
Her touching story prompted members of the audience to also add their own stories into the mix.
Freda was given the gift of Myozyme through Genzyme/Sanofi’s compassionate access program in 2017. This followed years of requests for treatment being denied by PHARMAC.
Ronelle Baker
Muscular Dystrophy Association of NZ
 Chief Executive of Muscular Dystrophy Association of New Zealand shared insights, information and made a plea for collaboration within all communities affected by rare diseases to connect and work together for better outcomes for all. She touched on the future focus of MDANZ and the desire for collaboration and community leadership across multiple organizations moving forward.
Chief Executive of Muscular Dystrophy Association of New Zealand shared insights, information and made a plea for collaboration within all communities affected by rare diseases to connect and work together for better outcomes for all. She touched on the future focus of MDANZ and the desire for collaboration and community leadership across multiple organizations moving forward.
MDANZ offer amazing support and research within the field of muscular dystrophy. Ronelle discussed the changes that have occurred in their organization in the past year. Things like amending their Vision to “Freedom beyond limits” the Mission to “promoting freedom of choice in a responsive society” to make them simpler to understand.
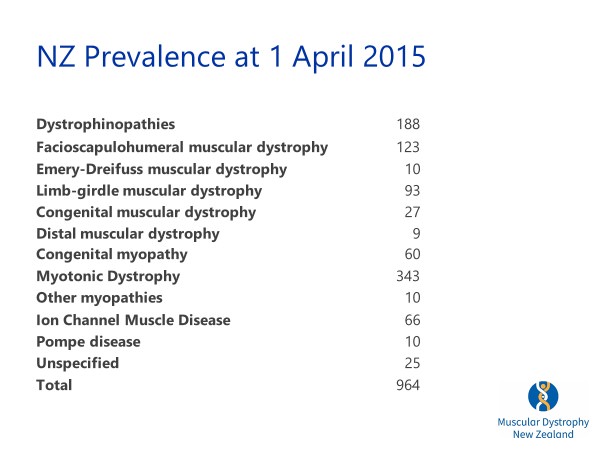
It is estimated that 4,500 New Zealanders have a neuromuscular condition. MDA supports patients with nearly 60 different conditions of which some are very rare. She outlined the conditions they cover and the prevalence of each.














Dr Dianne Webster
Auckland DHB
 Dr Webster spoke on the process and pitfalls that will be encountered when trying to have Pompe Disease included in the Newborn Screening program. She outlined the current screening process and the fact that the purpose of screening is to achieve health gains. If systems are not in place to add benefit to the patient after diagnosis then is there actually any benefit achieved? Questions were raised as to what may come from early diagnosis or if people were carriers, would that create false positives?
Dr Webster spoke on the process and pitfalls that will be encountered when trying to have Pompe Disease included in the Newborn Screening program. She outlined the current screening process and the fact that the purpose of screening is to achieve health gains. If systems are not in place to add benefit to the patient after diagnosis then is there actually any benefit achieved? Questions were raised as to what may come from early diagnosis or if people were carriers, would that create false positives?
Dr Diane Webster, clinical scientist at LabPLUS at Auckland DHB spoke about new-born screening and the complexities of ethics when considering the addition of a new rare disorder to newborn screening. The process and policy framework of adding conditions to newborn screening was explained which involves a wide range of information being considered including looking at what other counties are doing. Questions include: Is there a better way to do this? Just because we can screen, do we? Does testing need to occur before symptoms develop or just once symptoms develop? Is there an effective treatment available that works? This process then follows a framework through a governance group (including a technical group) and national screening advisory committee to allow decisions based on overall health gains with the focus of getting more benefit than harm for the population. Pompe patients can fill out a new disorders form which will initiate this process and were encouraged to do so.
Dr Webster explained that screening can lead to positives when the person would not go on to get the full disease and this can cause avoidable mental distress. Other ethics relate to the right to know for the child and that when an identified family have a second baby genetic testing is not always done as it is not to the babies’ benefit. Other factors mentioned included mass screening versus diagnostic screening, and only offering screening for family studies with a known patient, along with a focus on increasing clinical awareness for differential diagnosis rather than screening. This raises emotive issues as once symptoms develop the child has potentially already suffered irreversible damage and ERT or gene therapy benefits may be limited.
Dianne played devil’s advocate about newborn screening. Was it a good thing? Would it change the way parents treat children with LOPD who may not develop symptoms until their later years?
- A question I have is, if there is no known history, as there usually isn’t with an autosomal recessive disorder, would screening be done if it was only offered for people with a known history of the disease? If no, then we will miss the people with Pompe disease.






































Raymond Saich
Australian Pompe Association
 The president of the Pompe Association in Australia Raymond Saich lobbied Federal Government to place Myozyme for Pompe disease on the Pharmaceutical benefits scheme. He was awarded a Medal of the Order of Australia in the Queen’s Birthday honours list for this work.
The president of the Pompe Association in Australia Raymond Saich lobbied Federal Government to place Myozyme for Pompe disease on the Pharmaceutical benefits scheme. He was awarded a Medal of the Order of Australia in the Queen’s Birthday honours list for this work.
Raymond explained how his “pleasantly persistent” campaign to the Australian Federal Government eventually led to success for Pompe patients. He was able to locate champions within government who cared for this issue and create momentum with 136 minister letters, 30 articles in the media, print or on TV and two speeches made in parliament. The huge difference to New Zealand is that they have politicians who are willing to support patients.
Raymond said they had to fight perceptions that it was an old person’s disease. So all meetings with politicians were conducted using a team of three people, one being a young patient or mother so that the clarity of how this disease affects the range of ages was understood and appreciated.
Myozyme is available in 76 countries, however in New Zealand this drug is not available to adult patients although it is funded for new-borns with Infantile Onset Pompe Disease (IOPD). Currently there are no babies in NZ with this disease, and no new-born screening. Without screening for early diagnosis and subsequent treatment, the outcome is gloomy for any clinically identified babies as by the time symptoms are identified and the system of diagnosis has run its course vital treatment time is lost and the infant would be likely to die before diagnoses.
















Miriam Rodrigues
Muscular Dystrophy Association of NZ
 Miriam provided an overview of the NZ Neuro Muscular Disease Registry which has a primary aim of enabling participation in research, including clinical trials and natural history studies, and a secondary aim of obtaining molecular confirmation of diagnoses. She gave an indication of the numbers of people involved and that they were spread across the country.
Miriam provided an overview of the NZ Neuro Muscular Disease Registry which has a primary aim of enabling participation in research, including clinical trials and natural history studies, and a secondary aim of obtaining molecular confirmation of diagnoses. She gave an indication of the numbers of people involved and that they were spread across the country.
It was stated that many people diagnosed with Limb Girdle Muscular Dystrophy probably actually have Pompe disease as only 14 out of 59 people have a molecular diagnosis for LGMD.
The future direction for the registry is to facilitate diagnosis, continue to contribute data to natural history and feasibility studies, assist in recruitment for trials and studies, and advise researchers.
Miriam spoke about the Neuromuscular Disease Registry and she urged all Pompe people to sign up with it so that we can gain a better understanding of our needs.
To sign up to the registry, please visit this website:
http://www.mda.org.nz/Our-Research/NZ-NMD-Registry
Annelise La Roche
Resmed
 Annelise is the Business Manager for ResMed.
Annelise is the Business Manager for ResMed.
Annelise spoke on the benefits that may be available for patients using the sipper machine.
She demonstrated all of the functions and options available and then made herself available for people to see the machine in action.
Open circuit mouthpiece ventilation: Concise clinical review











Anthony Earp
Sanofi
 Anthony briefed the audience on the Sanofi Genzyme Humanitarian Programs around the world and how it has provided a lifesaving stopgap between patients being diagnosed and an established patient care support system being implemented in their country. It is a program that can serve patients for short term or long term depending on the individual’s requirements. It has been going for over 25 years and there are no plans to stop it.
Anthony briefed the audience on the Sanofi Genzyme Humanitarian Programs around the world and how it has provided a lifesaving stopgap between patients being diagnosed and an established patient care support system being implemented in their country. It is a program that can serve patients for short term or long term depending on the individual’s requirements. It has been going for over 25 years and there are no plans to stop it.
They are currently supporting over 700 patients around the world with Gaucher, Fabry, Pompe, MPS I and MPS II with plans to include ASMD.
Anthony talked about bringing expensive drugs to people in countries where they are not funded. This also applies to New Zealand as 4 Pompe patients were given the gift of Myozyme because of lack of funding by successive governments over the years.







Michelle Hackenberry
Data Registry Services
 Michelle spoke on Data Registry Services whose mission is to enhance patient, physician, and pharmaceutical comprehension of disease and associated treatments through increased data precision and patient participation in medical data registries.
Michelle spoke on Data Registry Services whose mission is to enhance patient, physician, and pharmaceutical comprehension of disease and associated treatments through increased data precision and patient participation in medical data registries.
The registry is supported by Sanofi Genzyme and participants are provided free access to all of their medical records, which is particularly useful for people who are travelling and require access to their information in an emergency. The registry stores the details of patients with Pompe, Gaucher, Fabry or MPS on a voluntary basis.
This registry collects information along a similar line as the Neuromuscular registry in NZ but this one is specifically for Pompe. Work is being done so that NZ patients can join the registry at Data Registry Services.







Anthony Earp
Sanofi
Anthony returned to the lectern to explain the process for bringing a rare disease drug to the patient. He covered some of the difficulties faced; even working out what is a rare disease is, is different all around the world. In New Zealand there isn’t an official definition of what is rare.
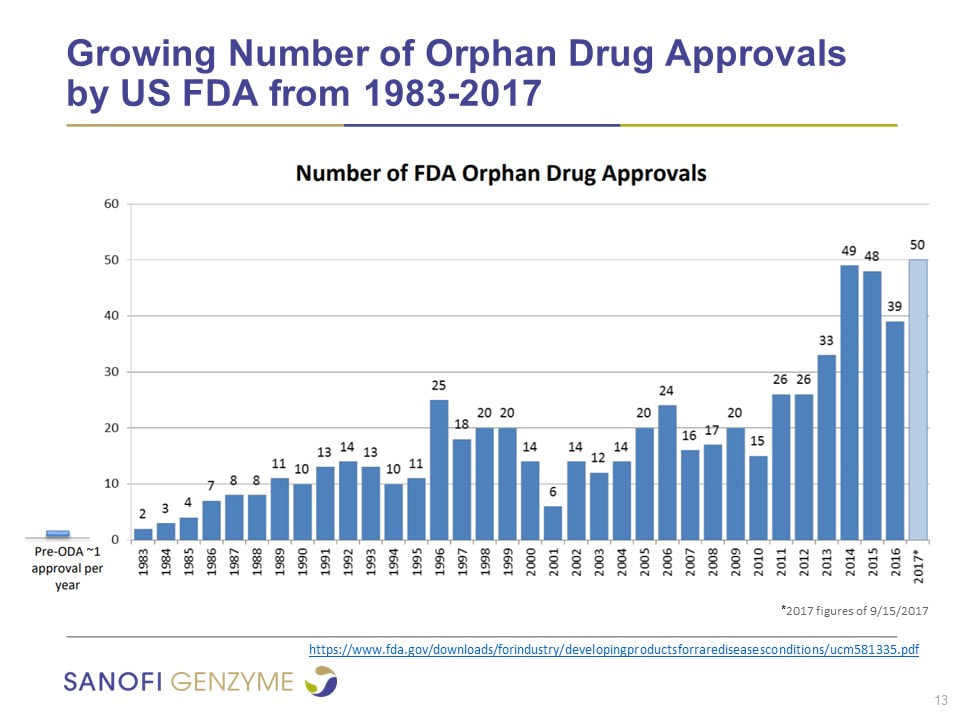
Drug companies do deals with governments to obtain market exclusivity. This exclusivity can vary from country to country and vary in duration. Advances in technology have encouraged more companies to pursue rare disease treatments in the past decade.
Since many rare diseases have a genetic component or cause, greater knowledge in this area is progressing therapies significantly. Some of the challenges that arise are the small number of people available for studies/trials, a sick and deteriorating patient population, the length of time to achieve a measurable effect, and diversity in regulations across different jurisdictions. There are also a lot of specific paediatric challenges as often the gene effect is the same in children as in adults however the effect of the disease on children can be much more severe, so treatment needs to be initiated early in order to prevent irreversible end-organ damage. Genzyme has invested more than $1billion in manufacturing facilities around the world as manufacturing ERTs is a highly complex, resource-intensive and time-consuming endeavour.





























Dr Barry Byrne
University of Florida
Dr Byrne discussed the difference between ERTs and Gene Therapy and the possible way ahead for treatment of Pompe disease
A gene therapy that treats respiratory problems in early-onset Pompe disease was shown to be safe during its first human trials.
The therapy uses a harmless adeno-associated virus to deliver a functional copy of the affected gene to muscle cells in the diaphragm of patients who have respiratory troubles. Nine patients completed a trial that found the therapy agent produced no adverse effects and improved respiratory function in the study participants.
The safety finding follows earlier publications by UF Health researchers that established their gene therapy’s effectiveness: Breathing function improved in a group of nine patients when compared with participants whose treatment involved breathing exercises.
Dr Byrne said the gene therapy represents a significant advance over enzyme replacement therapy, which involves regular intravenous infusions that contain the needed enzyme. While the enzyme treatment can reduce symptoms and prevent long-term damage, it is expensive and does not address the neurological aspects of Pompe disease. Current enzyme replacement treatments cannot cross the blood-brain barrier.
Restoring gene function in neurons, or nerve cells, is particularly important because it affects muscle cell functioning and signalling. We know the gene therapy vector — when given systemically — actually enters the brain and corrects neurons that have the same deficits as muscle cells in Pompe disease.
In the recent study, patients were assigned to two dosing groups and treated with different amounts of the gene therapy. Neither group suffered any adverse reactions that were attributed to the therapy.
One issue that will continue to be studied is the gene therapy’s durability. Byrne said a child’s growth will likely reduce the effect of the treatment, meaning that it could last five to 10 years in school-age children and probably less if given to an infant, unless there is a provision for later exposure to the therapy.
Next, researchers will begin work on a trial to test the gene therapy systemically by administering it intravenously. The National Institutes of Health has provided a three-year grant and patient enrolment should begin shortly. By giving the treatment intravenously, researchers expect to treat most of the disease-affected tissues, including neural tissues, the heart, the diaphragm and the peripheral muscles. They also will use immunosuppressive drugs that prevent potential reactions to the treatment and would allow for repeat administration in the future. That approach will address the limited durability of the treatment, researchers said. The study will be conducted with collaborators at the NIH.
Chelsea Karbocus and Dr Doug Laidlaw
Audentes Therapeutics


Chelsea and Doug came from the USA and introduced the audience to Audentes Therapeutics and the therapies they are currently working on developing. They have multiple products in development, targeting diseases that have little or no treatment options currently. It is hoped that human trials will be starting in 2019 for gene therapy for Pompe disease.
Chelsea discussed the patient advocacy policies and explained that integration of the patient and family perspective is at the core of their work.
Doug provided an overview on how a gene therapy approach works and how they are attempting to create treatments using a virus to deliver functional genes to the body.
We would be happy to have you direct folks to our website, where they can find the latest public information in our company press releases (see screenshot attached for navigation). https://www.audentestx.com/

We will also be updating the “patients + families” section in the next few months with some educational content, links to helpful resources, and of course our latest news as it relates to the Pompe program.
In addition, feel free to share either my contact information or the Patientadvocacy@audentestx.com email if anyone would like to reach out with further questions.
Thank you very much,
Chelsea
Comments from attendees:
- I just wanted to drop you a quick note to thank you for the excellent meeting over the weekend. Not only was the content excellent but I have to compliment you also on the warmth and positive tone of the whole event. As a newcomer to your community I felt completely welcome. Good luck with all of your efforts on behalf of people dealing with Pompe disease and I look forward to working with you and the team in the future.
- The conference was a huge achievement. Well done to everyone involved.
- Relaxed but informative.
- Knowledgeable speaker list.
- Very good speakers and a good mix of patient and clinical focus.
- The program was well balanced covering a variety of topics of interest.
- Meeting was very well organized and buttoned up!!! Great job.
- Great opportunity and quality; loved the goodie bag – but mainly enjoyed the networking opportunity. Well done!
- Good selection of speakers nationally and internationally.
- Congratulations on a great meeting and thank you for putting so much time into it.
- Thank you for a very successful first meeting!
- Excellent goodie bags and range of resources 🙂
- It was great to be part of it and I hope there’ll be more of these meetings to come!
- Today was AWESOME! Well done! And please pass on my congratulations to everyone else who helped organise – it really was such a good meeting. It was great too to meet the Byrnes and it turns out there are common interests across a number of different areas so I hope they’ll be back. Hope you’re taking a break over the next little while – you deserve it!
- Huge congratulations on your magnificent first ever Pompe NZ Network Conference on Friday and Saturday last week! We were so impressed with the professionalism and organisation that had gone into the event and the unity and feeling of Whanau it generated. We could have learned much from you in organising our little meeting!! Thank you also for inviting me to speak and giving me the gift of hearing the extraordinary Barry Byrne speak about his work. With him on the case, Pompe is in the best possible hands. His science is impeccable and above and beyond what I could have ever dreamed to accomplish as a scientist in this country. I was left with a sense of joy and one-ness with your Pompe family and also a sense of frustration at the lack of progress in this country on your hugely deserving part.
Speaker Biographies
Dr Barry Byrne, M.D., Ph.D.
Academic Title
Director, UF Powell Gene Therapy Center
Professor, Pediatrics and Molecular Genetics & Microbiology
Associate Chair, Pediatrics
Dr. Barry J. Byrne is a clinician scientist interested in a variety of rare diseases, with specific attention to developing therapies for inherited muscle disease. As a pediatric cardiologist, his focus is on conditions that lead to skeletal muscle weakness, cardiac dysfunction and respiratory dysfunction. His research team has made significant contributions to the understanding and treatment of Pompe disease, a type of muscular dystrophy resulting from abnormal glycogen accumulation in the muscle. His current research has focused on developing new therapies using the missing cellular protein or the corrective gene to restore muscle function in Pompe and other inherited myopathies.
Dr Byrne is the Associate Chair of Pediatrics and Director of the Powell Gene Therapy Center at the University of Florida. After obtaining a B.S. degree in Chemistry from Denison University, he pursued his medical education, as well as a Ph.D. in Microbiology and Immunology, at the University of Illinois. He completed his pediatric residency, cardiology fellowship training and post-doctoral training in Biological Chemistry at Johns Hopkins University. Joining the University of Florida in 1997, he has served in a variety of clinical, research and educational roles, and is now the Earl and Christy Powell University Chair in Genetics.
Key Publications
Found at Pubmed: http://www.ncbi.nlm.nih.gov/sites/entrez under “Byrne BJ”[Author]
Tagged as: Autosomal recessive lysosomal storage disease, Barth Syndrome, Cardiomyopathy, Cellular and Molecular Therapy, Clinical Trials, Gene therapy, Genetic diseases, Hemophilia, Inherited muscle disease, Muscular dystrophy, Pompe Disease
Raymond Saich OAM – President Australian Pompe Association
A long-time advocate for sufferers of Pompe disease, Raymond used his position as president of the Australian Pompe’s Association to lobby the federal government to place valuable medication onto the Pharmaceutical Benefits Scheme (PBS). It’s for this work that he has been awarded a Medal of the Order of Australia in the Queen’s Birthday Honours List.
Raymond also has Pompe disease.
Michelle Hackenberry – NR-CMA
Michelle Hackenberry is a Nationally Registered Certified Medical Assistant. She is the Study Coordinator/Chief Operations Officer with Data Registry Services based in Pittsburgh, PA., a company Michelle and her business partner, Eric Rice, founded in 2014.
Together with their PI, Dr. Michael Joseph, they recognized a need for registry site availability for those affected with lysosomal storage diseases who were not actively being treated by a physician at a registry site.
Michelle also is a proud mom to her 3 adult children; two of which are affected by juvenile onset Pompe Disease.
Dr Collette Bromhead – Chief Executive NZORD
Collette is a clinical scientist with 20 years’ health sector leadership and academic experience. She comes to NZORD from Massey University where her research has focused on improving the performance and accessibility of genomic tests. Collette has a strong background in health advocacy and has held multiple advisory roles for the Ministry of Health.
With a career that bridges both the sciences and clinical medicine, Collette enjoys bringing ideas from these fields together to increase the understanding of patients, clinicians and colleagues.
Annelise La Roche – Business Manager for ResMed
Annelise has been with the company for 3 1/2 years and supports New Zealand healthcare professionals and patients to ensure they have access to the latest products and technologies from ResMed. She has worked for many years within the technology health sector of New Zealand, she is a Registered Nurse, previously specialising in Paediatrics , Midwifery and Neonatal Intensive Care medicine. She has worked in New Zealand & overseas. Her hobbies include activities that involve unique outdoor experiences & meeting a wide range of people with different fields of interest.
Ronelle Kiterangi Baker – Chief Executive MDANZ
Following a varied 20 year career that includes DHB and NGO settings, Ronelle became the first CEO of the Muscular Dystrophy Association of New Zealand to have lived experience of a neuromuscular condition in February 2016. It is therefore no surprise that one of her favourite leadership quotes is by former US pro basketball coach John Wooden – “The most powerful leadership tool you have is your own personal example.”
Ronelle will share insights and information with delegates, focusing on the power of support groups working in collaboration to achieve better outcomes.
Anthony Earp – Senior Medical Manager at Sanofi Genzyme
Anthony Earp is a pharmaceutical industry professional with over 25 years’ experience.
The vast majority of this time has been spent at Genzyme (now Sanofi Genzyme) working in the field of rare diseases, in particular the lysosomal storage diseases and endocrine cancers.
Anthony currently holds the title of Senior Medical Manager.
Chelsea Karbocus – Senior Manager, Patient Advocacy & Engagement at Audentes Therapeutics
Chelsea Karbocus joined Audentes Therapeutics as Senior Manager, Patient Advocacy & Engagement in February 2018. She has more than 8 years of experience in patient engagement and communications.
Chelsea has had the opportunity to partner with and learn from patients, families, and patient advocacy organizations across many different therapeutic areas. She has extensive experience developing patient education materials, communication strategies, and integrating the patient and family perspective into both clinical and commercial activities.
Chelsea is honoured to partner with the patient community in an effort to make a meaningful impact for patients and their families.
Dr Doug Laidlaw – Senior Medical Science Liaison at Audentes Therapeutics
Doug Laidlaw, PhD is currently a Senior Medical Science Liaison in Medical Affairs with Audentes Therapeutics, supporting drug and clinical development, and education related to the company’s gene therapy development programs for X-Linked Myotubular Myopathy, Crigler-Najjar Syndrome, Pompe Disease and CASQ2-related Catecholaminergic Polymorphic Ventricular Tachycardia.
Prior to joining Audentes, Doug worked in many therapeutic areas, including CNS disorders, Chronic Kidney Disease, Multiple Sclerosis and in rare genetic disorders such as Fabry Disease and Polycystic Kidney Disease. He has close friends and family members with both rare and chronic diseases and is committed to supporting novel research that brings new therapies and hope to patients.
After completing undergraduate studies in Civil Engineering and in Health Sciences in the California State University system, Doug studied biomechanics at the University of Arizona and received a Master of Science in Exercise and Sport Sciences. He then completed a doctoral program in Physiological Sciences, focusing his research on understanding the mechanisms underlying changes in the neural activation of muscles with advancing age. Doug is married and has a son studying business in the U.S. and a daughter studying Marine Sciences in Australia.
Dr Dianne Webster – Clinical Scientist at LabPLUS at Auckland District Health Board
Dr Webster has responsibilities for newborn metabolic screening and has won an international award for excellence in global leadership in standards development.
Dr Webster has been active for many years in the establishment of both written and physical standards and initiated the International Society for Neonatal Screening (ISNS) lexicon and minimum data set some 20 years ago. She is a past chair of ISNS Standard Committee for Quality Assurance, and was involved in the development of the ISNS Dried Blood Spot Reference Preparation.
More recently she was appointed co-chair of a new ISNS Committee on Guidelines and Quality Assurance and co-chair of the CLSI-ISNS working group on terminology in newborn screening to undertake a project that will lead to a concise list of terms that should be used in newborn screening publications.
Laura Byrne – B.S., P.T.
Laura received her Bachelor of Science degree from Denison University and her P.T. degree from the University of Pennsylvania. She has extensive experience working in the areas of rehabilitation and sports medicine. She has used aquatic therapy in her practice and is certified in Polestar Pilates, GyrotonicÒ, and GyrokinesisÒ. Laura takes advantage of these exercise systems, as well as associated equipment, to increase body awareness and improve balance, posture, and function. She specializes in training patients to maximize function as part of recovery from injury or in long-term care of chronic conditions.

Thank you again to our wonderful sponsors!
